 |
Case Report
The paradox of size: A case of indolent oncocytic adrenocortical carcinoma and review of literature
1 Department of Internal Medicine, The University of Texas at Austin, Austin, TX, USA
Address correspondence to:
Sujata Ojha
Department of Internal Medicine, The University of Texas at Austin, Austin, TX,
USA
Message to Corresponding Author
Article ID: 100151Z10SO2025
Access full text article on other devices

Access PDF of article on other devices


How to cite this article
Ojha S, Abdullah Y, Pandey ON, Goodgame B, Kumar P, Kulkarni M. The paradox of size: A case of indolent oncocytic adrenocortical carcinoma and review of literature. J Case Rep Images Oncology 2025;11(2):20–24.ABSTRACT
Introduction: Oncocytic adrenocortical carcinoma (OAC) is a rare histologic variant of adrenocortical carcinoma. It is characterized by cells with abundant eosinophilic cytoplasm because of high mitochondrial content. Unlike conventional Adrenocortical carcinoma (ACC), which is typically aggressive, OAC often presents with a more indolent biological behavior. The paradoxical features of OAC make the diagnosis and management challenging.
Case Report: A 45-year-old female presented with weight loss and left-sided abdominal pain, with initial imaging revealing a large (24 cm) left adrenal mass. Imaging revealed that the mass had radiologic signs and necrotic features concerning for malignancy. She subsequently underwent a left adrenalectomy with en bloc pancreatectomy, splenectomy, and left nephrectomy along with paraaortic lymphadenectomy. Histopathology revealed classic oncocytic morphology with low Ki-67 index (2%) and absence of any lymph node involvement, despite venous and capsular invasion. Due to the venous invasion alone, the tumor met the Lin–Weiss–Bisceglia (LWB) criteria for malignancy. Additional molecular analysis revealed a favorable diagnosis by showing TP53 alterations, low tumor burden, along with microsatellite stability. Postoperatively, the patient recovered and was administered external beam radiation.
Conclusion: This case is an example of a rare subtype of ACC and highlights a clinical paradox. In patients with OAC, a large tumor size with necrosis may not always be associated with a poor prognosis. Additionally, OAC has unique diagnostic and management challenges due to its rare nature. However, surgery is still the mainstay treatment for OAC, with the role of mitotane in adjuvant therapy still being explored.
Keywords: Adrenal cortical carcinoma, Adrenal tumor, Lin–Weiss–Bisceglia, Oncocytic adrenocortical carcinoma
Introduction
Adrenocortical carcinoma (ACC) is a rare aggressive malignancy that originates from the adrenal cortex and has an annual incidence of approximately 0.7 to 2 cases per million people [1]. Conventional ACC often presents with rapid tumor growth, frequently producing overt hormonal syndromes and harboring a poor prognosis if diagnosed at advanced stages. Despite the rarity, ACC is associated with a poor prognosis because it is aggressive in nature and can remain asymptomatic until it progresses making early detection challenging. The clinical presentation of ACC can vary, with patient exhibiting hormonal syndromes, while others may remain asymptomatic [2]. Oncocytic adrenocortical carcinoma (OAC) is a rare histopathological variant of ACC characterized by tumor cells that have an abundant granular, eosinophilic cytoplasm due to high mitochondrial content [3]. Unlike ACC, OAC often exhibits an indolent course, although it does have malignant potential [4]. The Lin–Weiss–Bisceglia (LWB) criteria are commonly used to differentiate between benign and malignant oncocytic adrenal neoplasms. The criteria focus on features such as high mitotic rate, atypical mitoses, and venous invasion [5]. Due to the rarity of OAC standardized treatment protocols are lacking, however like ACC, surgical resection remains the primary therapeutic approach. Further, its diagnosis and management can be challenging due to the paradox of large tumor size and low proliferative markers.
Case Report
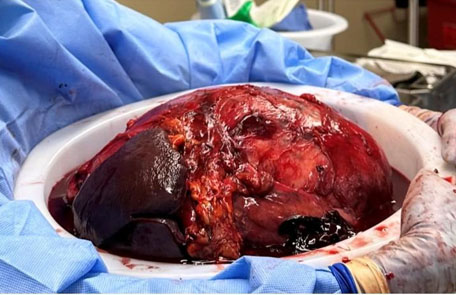
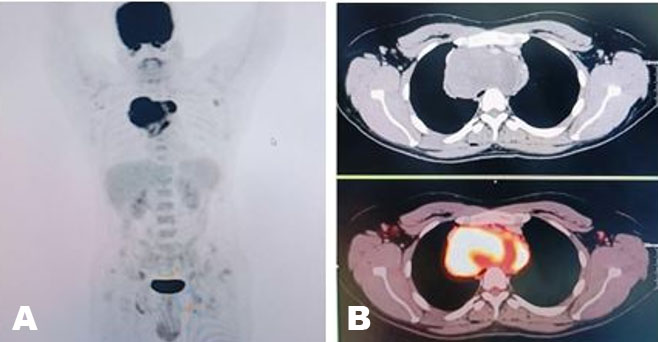
A previously healthy 45-year-old woman was admitted to the hospital for left-sided abdominal pain, significant weight loss of about eighty pounds over seven-month period, along with mild endocrine abnormalities. Pertinent physical exam findings included a large protuberant abdomen with palpable mass occupying the entire left upper quadrant; however, she did not have hirsutism, signs of virilization, or cushingoid features. Initial imaging revealed that she had a large left adrenal mass measuring 24.6 × 21.5 × 17.1 cm, with central necrosis and heterogeneous enhancement (Figure 1 and Figure 2). A 24-hour urinary free cortisol level was 74 μg/24 h, which indicated mild hypercortisolism. DHEAS was elevated at 325 μg/dL, suggestive of androgen overproduction without clinical signs of virilization. The urinary metanephrines were normal and normetanephrines were slightly elevated and measured at 1.5 nmol/L, indicating no evidence of pheochromocytoma. Additionally, the patient exhibited non-PTH-mediated hypercalcemia (total serum corrected calcium 11.4 mg/dL). Skeletal metastasis was ruled out with nuclear medicine bone scan. A multidisciplinary tumor board recommended an en-bloc surgical resection accounting for the size of the mass and radiographic features that were indicative of malignancy. The patient underwent a left adrenalectomy combined with a left nephrectomy, distal pancreatectomy, splenectomy, and extended lymphadenectomy via a chevron incision. The patient’s multiorgan resection successfully cleared the lesion, yielding negative margins and removing 54 lymph nodes, all pathologically negative. This underscores that even with large adrenal masses, nodal spread may be absent if the tumor’s proliferative index is relatively low. Early vascular control near the renal hilum was established to minimize hemorrhagic risk. The surgery was complicated by an estimated blood loss of 2.5 L, requiring intraoperative transfusions. Postoperatively, the patient received stress-dose hydrocortisone to mitigate the risk of adrenal insufficiency. After discharge, the patient was started on external beam radiation therapy for five weeks and identified for long-term surveillance. The patient did not receive adjuvant therapy with mitotane. Subsequent imaging completed three months after completion of radiation therapy revealed no evidence of tumor recurrence. Corrected serum calcium at the same time was 10.0 mg/dL. The patient is currently being closely followed up in the endocrinology clinic for steroid taper.
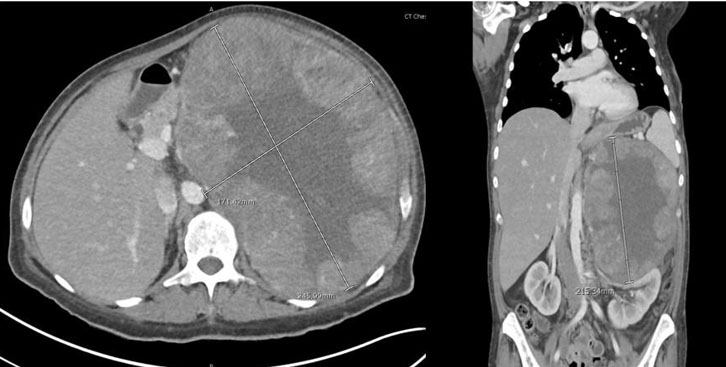
Discussion
Oncocytic adrenocortical carcinoma is a rare variant of ACC, characterized by abundant eosinophilic cytoplasm due to an increased number of mitochondria. This was first described in literature in 1986, by Kakimoti S et al. as a rare tumor of the adrenal gland [6]. Oncocytic adrenocortical carcinoma is identified as a separate entity due to the unique histopathological and molecular features. Due to its rarity, data on OAC is limited. However, this review aims to summarize the current literature on epidemiology, histopathological classification, molecular alterations, clinical behavior, and treatment strategies for OAC. Oncocytic adrenocortical carcinoma makes up less than 10% of all adrenocortical tumors and primarily affects adults, with the median age at diagnosis ranging from 40 to 60 years [6],[7]. Unlike conventional ACC, which shows a slight female predominance, OAC does not display a clear gender preference. The clinical presentation of OAC can vary widely; some patients experience symptoms from mass effect, such as abdominal pain or discomfort, while others show signs of hormonal overproduction, like Cushing’s syndrome or virilization [3]. However, OACs are generally less functional than conventional ACC.
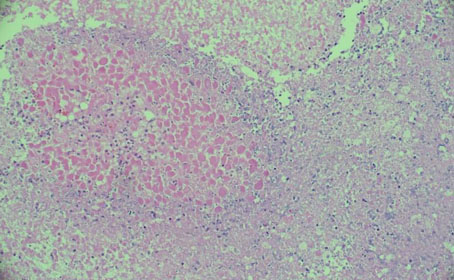
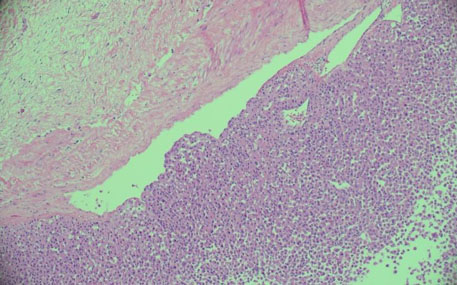
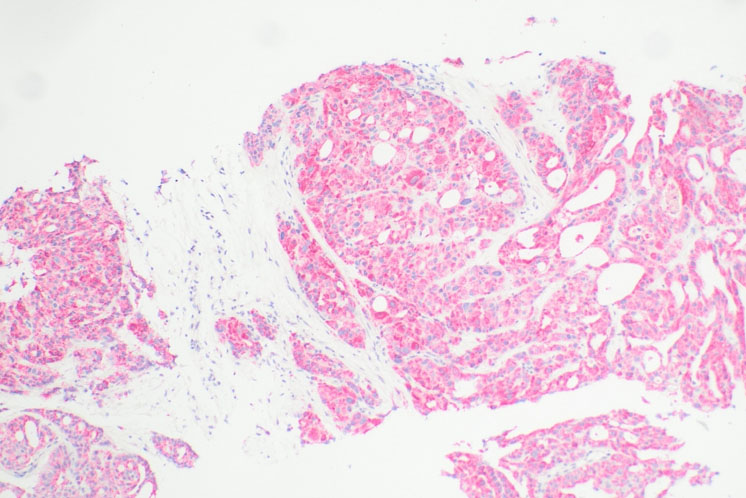
The histological characteristic of OAC is the presence of large polygonal cells with abundant eosinophilic cytoplasm, as a result of mitochondrial accumulation [3]. The diagnostic criteria used for ACC, the Weiss system, overestimates malignant potential in oncocytic tumors, therefore a separate classification system called the Lin–Weiss–Bisceglia (LWB) classification is used for oncocytic tumors [5],[8]. The LWB system divides oncocytic adrenocortical neoplasms (OANs) into benign, borderline, and malignant categories based on three main criteria: a mitotic rate greater than 5 per 50 high-power fields, vascular invasion, and metastasis. Malignancy is diagnosed if any of these criteria are present, while borderline tumors show only minor criteria, such as a tumor size greater than 10 cm, necrosis, or capsular invasion. This classification system has enhanced the ability to differentiate between benign oncocytic adenomas and OACs. In our patient, the resected specimen showed a well-circumscribed tumor weighing 5458 g, and measuring 26.0 × 25.0 × 16.0 cm. The tumor also had extensive hemorrhage and necrosis, along with a central fibrous scar [9]. Microscopically the patient’s tumor matched the classic histological findings for OAC and exhibited a low Ki-67 index of 2% suggesting a low proliferative rate. However, the tumor did exhibit vascular (renal vein) and capsular invasion (Figure 3, Figure 4, Figure 5, Figure 6). Based on the LWB criteria, malignancy was established due to the vascular invasion. Next-generation-sequencing (NGS) showed TP53 pathway alterations. The malignancy had a low tumor mutation burden and was found to be microsatellite stable. These findings are consistent with recent research suggesting a distinct mitochondrial metabolic profile with an oncocytic variant of ACC [4].
Molecular profiling studies have shown that OAC exhibits distinct genetic alterations compared to conventional ACC. While TP53 mutations are common in both subtypes, OACs have fewer alterations in genes like CTNNB1, IGF2, and ZNRF3, which are frequently involved in conventional ACC [1]. Literature suggests that oncocytic tumors may have unique mitochondrial metabolic profiles, with changes in genes related to oxidative phosphorylation and mitochondrial biogenesis [10]. The low tumor mutation burden and microsatellite stability of OAC further distinguish it from conventional ACC, indicating potential resistance to immune checkpoint inhibitors. However, due to the rarity of OAC, comprehensive genomic studies are still limited, and further research is needed.
Studies suggest that OAC has a more indolent course with a lower likelihood of distant metastasis, especially compared to ACC [3],[4]. Typically factors like large tumor size, high mitotic activity, and vascular invasion, are associated with a worse prognosis. However, despite being a variant of ACC, which is associated with a poor prognosis, OAC tends to have better survival rates, especially when complete surgical resection is achieved. The mainstay treatment for OAC is surgical resection, primarily due to the risk of invasion locally along with risk of recurrence [11],[12]. In high-risk conventional ACC, adjuvant mitotane therapy is standard, however its role in OAC is unclear due to lower recurrence risk and its distinct molecular profile [12]. In retrospective studies low Ki-67 index and negative lymph node involvement has been shown to have no benefit from mitotane. These findings align with the ADIUVO trial, which compared surveillance to adjuvant mitotane in low-grade localized ACC and found no benefit of adjuvant mitotane [13]. In terms of further management, local radiation is usually reserved for future recurrence since efficacy of radiation in early-stage disease with negative margins is inconclusive. However, in our patient she underwent local radiation due to the size of her tumor, vascular invasion, and risk for future recurrence. For OAC, surveillance is crucial due to the potential for late recurrence thus serial imaging and biochemical monitoring is recommended every 3–6 months for at least five years [14].
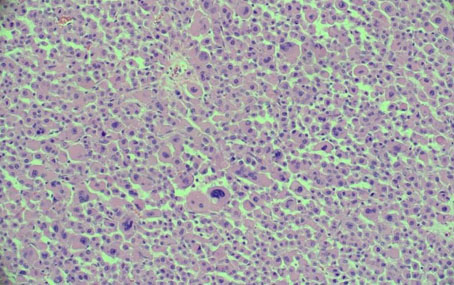
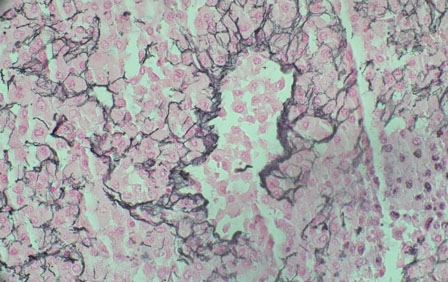
Conclusion
Oncocytic adrenocortical carcinoma represents a rare diagnostically challenging variant of ACC. While LWB classification has increased the ability to distinguish between malignant and benign forms, the question of the significance of genetic profile and post-surgical treatments remains. Although routine adjuvant mitotane or radiotherapy is typical for aggressive ACC, the specifics of OAC, including Ki-67 under 10%, negative margins, and an otherwise minimal endocrine overproduction, can justify a strategy towards imaging surveillance alone. The potential for late recurrence and metastatic spread is present for OAC, therefore lifelong surveillance is recommended. In summary, our case highlights that size and clinical appearance of a tumor is not always predictive of a poor outcome. This serves a reminder that pathology and clinical behavior do not always align.
REFERENCES
1.
Libé R, Huillard O. Adrenocortical carcinoma: Diagnosis, prognostic classification and treatment of localized and advanced disease. Cancer Treat Res Commun 2023;37:100759. [CrossRef]
[Pubmed]

2.
Wajchenberg BL, Albergaria Pereira MA, Medonca BB, Latronico AC, Campos Carneiro P, Alves VA, et al. Adrenocortical carcinoma: Clinical and laboratory observations. Cancer 2000;88(4):711–36.
[Pubmed]
3.
Kalra S, Manikandan R, Srinivas BH. Oncocytic adrenocortical carcinoma—A rare pathological variant. BMJ Case Rep 2015;2015:bcr2014208818. [CrossRef]
[Pubmed]
4.
Mills JK, Khalil M, Pasieka J, Kong S, Xu Y, Harvey A. Oncocytic subtypes of adrenal cortical carcinoma: Aggressive in appearance yet more indolent in behavior? Surgery 2019;166(4):524–33. [CrossRef]
[Pubmed]
5.
de Krijger RR, Papathomas TG. Adrenocortical neoplasia: Evolving concepts in tumorigenesis with an emphasis on adrenal cortical carcinoma variants. Virchows Arch 2012;460(1):9–18. [CrossRef]
[Pubmed]
6.
Kakimoto S, Yushita Y, Sanefuji T, Kondo A, Fujishima N, Kishikawa M, et al. Non-hormonal adrenocortical adenoma with oncocytoma-like appearances. Hinyokika Kiyo 1986;32(5):757–63.
[Pubmed]
7.
Duregon E, Volante M, Cappia S, Cuccurullo A, Bisceglia M, Wong DD, et al. Oncocytic adrenocortical tumors: Diagnostic algorithm and mitochondrial DNA profile in 27 cases. Am J Surg Pathol 2011;35(12):1882–93. [CrossRef]
[Pubmed]
8.
Urusova L, Porubayeva E, Pachuashvili N, Elfimova A, Beltsevich D, Mokrysheva N. The new histological system for the diagnosis of adrenocortical cancer. Front Endocrinol (Lausanne) 2023;14:1218686. [CrossRef]
[Pubmed]
9.
Hodgson A, Pakbaz S, Mete O. A diagnostic approach to adrenocortical tumors. Surg Pathol Clin 2019;12(4):967–95. [CrossRef]
[Pubmed]
10.
Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim Biophys Acta 2011;1807(6):633–42. [CrossRef]
[Pubmed]
11.
Kanitra JJ, Hardaway JC, Soleimani T, Koehler TJ, McLeod MK, Kavuturu S. Adrenocortical oncocytic neoplasm: A systematic review. Surgery 2018;164(6):1351–9. [CrossRef]
[Pubmed]
12.
Mihai R, De Crea C, Guerin C, Torresan F, Agcaoglu O, Simescu R, et al. Surgery for advanced adrenal malignant disease: Recommendations based on European Society of Endocrine Surgeons consensus meeting. Br J Surg 2024;111(1):znad266. [CrossRef]
[Pubmed]
13.
Terzolo M, Fassnacht M, Perotti P, Libé R, Kastelan D, Lacroix A, et al. Adjuvant mitotane versus surveillance in low-grade, localised adrenocortical carcinoma (ADIUVO): An international, multicentre, open-label, randomised, phase 3 trial and observational study. Lancet Diabetes Endocrinol 2023;11(10):720–30. [CrossRef]
[Pubmed]
14.
ENS@T – adrenocortical carcinomas (acc). [Available at: https://ensat.wildapricot.org/page-1317312]
SUPPORTING INFORMATION
Acknowledgments
Artificial Intelligence, ChatGPT 4.0, was utilized for grammar and language refinement. AI-generated data or results are not included in this manuscript. All scientific interpretations are completed by the authors.
Author ContributionsSujata Ojha - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Youssef Abdullah - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Om Narayan Pandey - Acquisition of data, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Boone Goodgame - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Pratima Kumar - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Mrinalini Kulkarni - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guaranter of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthors declare no conflict of interest.
Copyright© 2025 Sujata Ojha et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}